Advanced HPC Based Drug Discovery with converged Deep Physics and AIC
Presentation of the problem and objective of the experiment
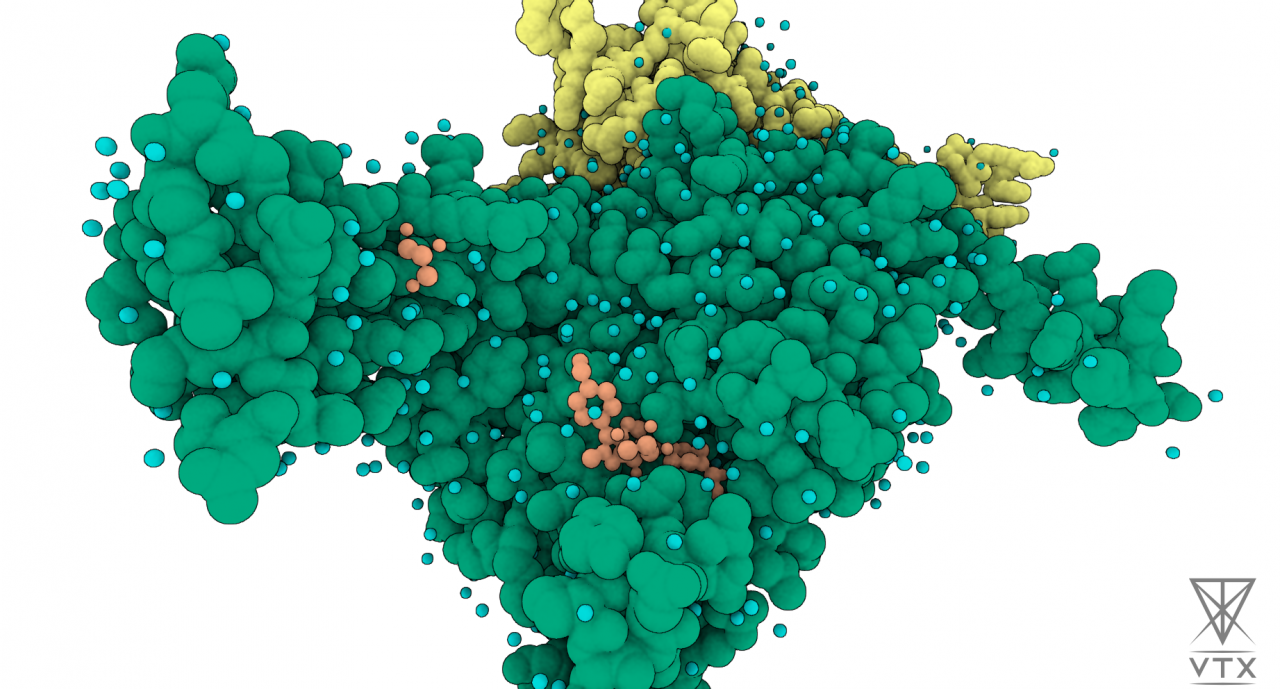
As time constraints and agility become crucial in the pharmaceutical industry, especially in the early stages of the drug discovery process, development of a consolidated and unified HPC framework paves the way for a major breakthrough. Such a framework will dramatically reduce the structure generation time, binding free energy calculation time, and computing costs, allowing faster delivery of a collection of optimally designed candidate drugs to their business partners. With this framework, we aim to reduce drug discovery time by 25% and costs by 20%.
Short description of the experiment
The goal of this experiment is to synergistically combine the expertise in the high-end calculation of absolute binding free energy of protein-ligand complex developed by Qubit Pharmaceuticals and Iktos’s AI-based deep generative modeling algorithms for novel compound generation and lead optimization. The combination of physics-based simulation and AI-driven deep generative modeling algorithms may lead to the development of a highly efficient drug discovery technology.
Given their technical expertise and domain applicability, the scaffold hopping approach is a well-suited case study for both companies to develop and explore commercial opportunities in the early drug discovery arena. Specifically, the new technology aims to discover structurally novel compounds starting from known actives by modifying the central core structure of the molecule.
UPDATE:
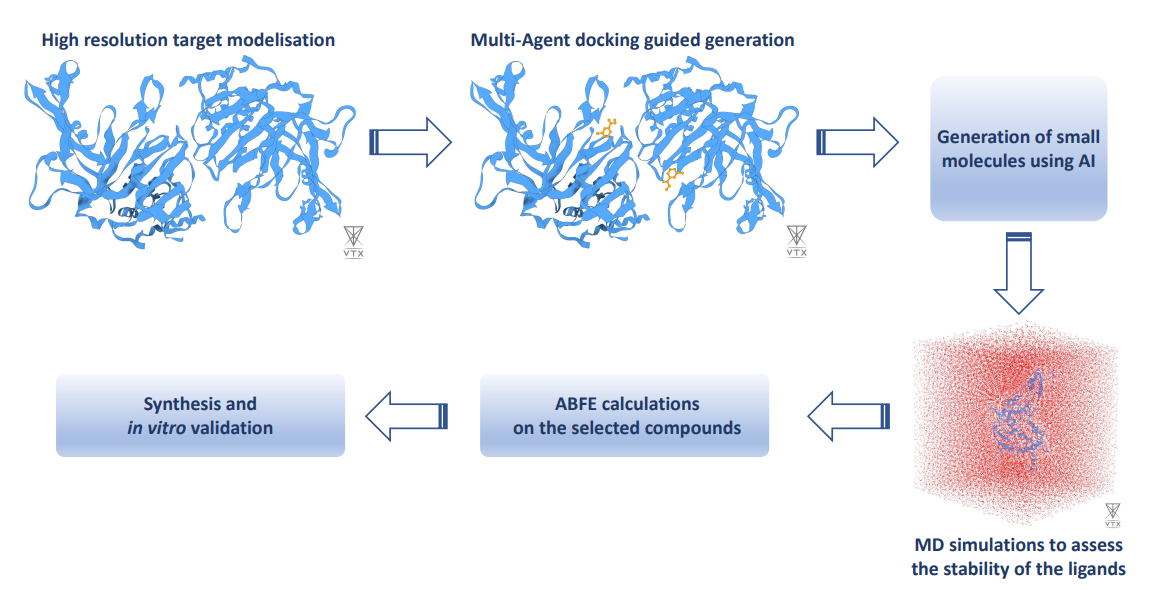
In this experiment, the expertise in Molecular Dynamics (MD) based approaches developed by Qubit Pharmaceuticals is combined to Iktos expertise in compound generation and lead optimization using AI algorithms. This collaboration will lead to the design of entirely new compounds, while simultaneously optimizing the chemical properties towards drug-like molecules.
In the context of this experiment, partners performed absolute binding free energy calculations on a series of protein-ligand complexes to validate the designed workflow dedicated to modeling the novel studied protein. The results show a high correlation between the computed absolute binding free energies and the corresponding experimental binding affinities.
Currently, partners are generating new small molecules using AI-based deep generative modeling algorithms. Next, they will evaluate the stability of those compounds using an MD-based approach. This will enable them to select a subset of potential compounds. Then, the validated absolute binding free energy pipeline will be used to assess the affinities of the selected compounds. Based on the computed affinities, partners will synthesize and test the most promising molecules in vitro. This hybrid MD - AI workflow will dramatically reduce time to the solution from target validation to drug candidate nomination.
Organisations involved:
End User: Iktos
Domain Expert: Qubit Pharmaceuticals